Hemofagocytair syndroom
Auteurs: Ilse Kuipers, Erfan Nur
Inleiding
Hemofagocytaire lymfohistiocytose (HLH), ook wel het hemofagocytair syndroom genoemd, is een potentieel levensbedreigende aandoening die gekenmerkt wordt door ongecontroleerde immuun activatie en weefselschade. Gezien de hoge mortaliteit, moet er bij verdenking HLH snel gehandeld worden. Er wordt gestreefd om de diagnostiek binnen 24 uur in te zetten (zie tabel 2) en indien een patiënt voldoet aan de criteria voor HLH (zie tabel 3) moet behandeling gestart worden, soms zelfs voor afronding van de diagnostiek naar de onderliggende oorzaak. Ook bij een sterke verdenking op HLH, waarbij er nog niet aan 5 van de 8 criteria wordt voldaan, wordt gestart met therapie.1 De HLH-probability calculator (H-score), ontwikkeld op basis van retrospectieve data van volwassenen, kan hierbij behulpzaam zijn (http://saintantoine.aphp.fr/score/) (sensitiviteit 93% en specificiteit 89%).
Pathogenese
In de normale situatie leidt een virus of maligniteit onder andere tot activatie en proliferatie van T en natural killer (NK) cellen, macrofagen en productie van cytokines (Figuur 1A). Cytotoxische T en NK cellen bevatten granules met perforine en granzyme. Via een gecoördineerd proces van transport, priming en binding wordt de inhoud van deze granules in contact gebracht met de membraan van de targetcel. Perforine veroorzaakt een porie waardoor granzyme de cel binnen dringt en leidt tot celdood. Op deze manier wordt de geïnfecteerde (of kwaadaardige cel) opgeruimd, neemt de antigeen stimulatie af en dooft de immuunrespons uit. In het figuur zijn de eiwitten belangrijk in het cytotoxische proces beschreven die als het gevolg van een homozygote mutatie bij familiaire HLH ontbreken en dus dysfunctie van dit proces veroorzaken.

Bij HLH is er een afname van de granule-gemedieerde cytotoxische activiteit waardoor een negatieve feedback ontbreekt en er een accumulatie van geactiveerde T-lymfocyten en macrofagen in weefsels ontstaat (figuur 1B). Een belangrijk onderdeel in de pathogenese is de ongecontroleerde productie van interferon-γ. Via stimulatie van de macrofagen veroorzaakt dit een cytokinestorm.
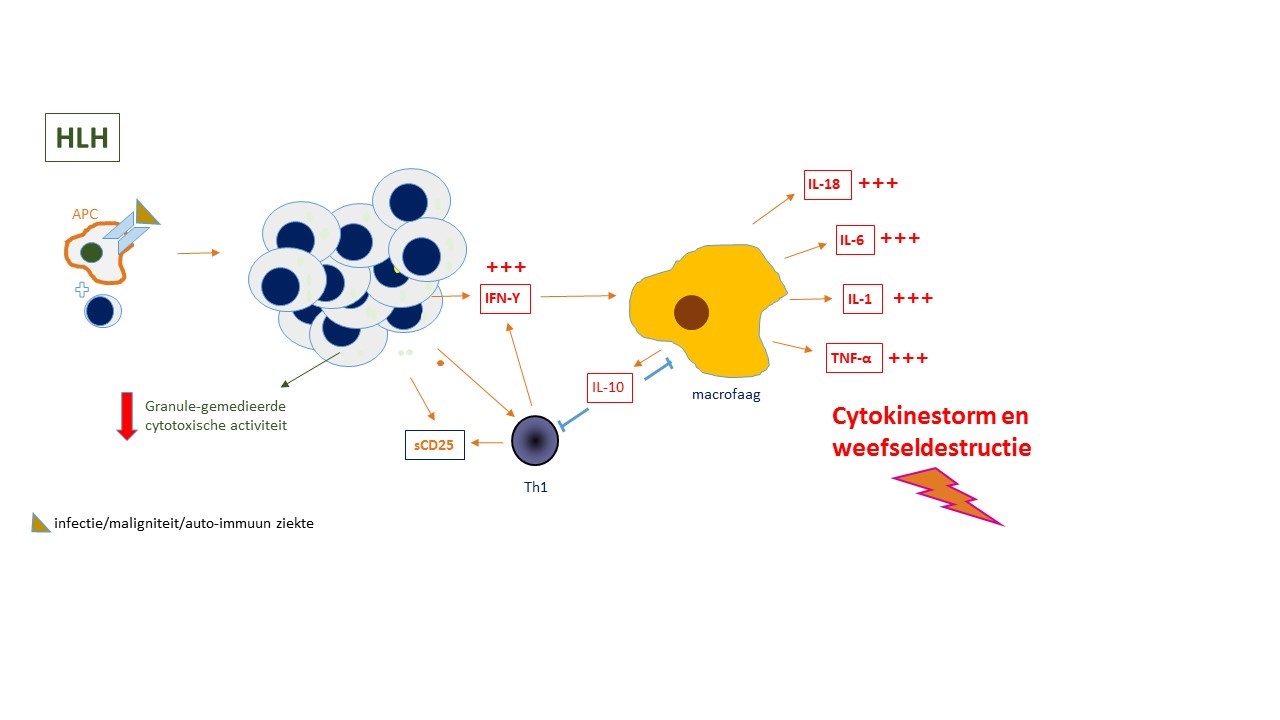
Oorzaken
Er bestaan 2 vormen; de primaire (hereditaire) vorm en secundaire HLH. De primaire vorm wordt veroorzaakt door mutaties die leiden tot een dysfunctie van de cytotoxische T en natural killer (NK)-cellen en wordt vooral op kinderleeftijd gezien. De geschatte incidentie van dit ziektebeeld bij kinderen is 0,1-1 per 100.000 per jaar.2 Secundaire HLH heeft hetzelfde fenotype en wordt uitgelokt door een infectie, maligniteit, auto-immuunziekte of therapie. Soms wordt er geen uitlokkende factor gevonden. De jaarlijkse incidentie wordt geschat op 1 per 800.000 volwassenen.2
De term macrophage activation syndrome (MAS-HLH) wordt gebruikt voor de subset van patiënten met HLH met als onderliggende trigger een auto-immuun aandoening (Still’s disease, lupus, vasculitis enz)
Een lijst van gevonden oorzaken/uitlokkende factoren van secundaire HLH in 2197 volwassen patiënten wordt in tabel 1 weergegeven.2 Zie ook het artikel van Rosado et al voor een lijst van HLH-geassocieerde aandoeningen.3
| Infecties N=1108 |
viraal (762): (oa EBV, HIV, herpes virussen, CMV, virale hepatitis, influenza, parvovirus B19) bacteriën (206): (oa TBC, rickettsia, S. Aureus, E. coli, atypische bacterien) parasieten (53): (oa Leishmaniasis, plasmodium, toxoplasma) schimmels: (37): (oa histoplasmose, aspergillus) ander virus/bacterie/parasiet of schimmel (50) |
| Maligniteiten N=1047 |
hematologische maligniteiten (981): T cel of NK cel lymfoom, B-cel non-Hodgkin lymfoom, Hodgkin lymfoom, Castleman’s disease, leukemie, MDS solide maligniteiten (32) ander niet gespecificeerd neoplasma (34) |
| Auto-immuunziekten N=276 |
SLE, Adult onset Still’s disease, RA, vasculitis, IBD, |
| Anders N=184 |
transplantatie (95): niertransplantatie, stamceltransplantatie, anders (76): medicatie (chemotherapie), chirurgie/biopsie, vaccinatie, trauma, diabetes/chronische leverziekte, zwangerschap, hemodialyse onbekend (13) |
Klinisch beeld
Het klinisch beeld van HLH wordt gekenmerkt door aspecifieke hyperinflammatie. In een retrospectieve analyse van 2197 HLH casus worden hoge koorts (96% van de patiënten) en hepatosplenomegalie (67-69% van de patiënten) als meest kenmerkende klinische bevindingen gevonden.2 Ook werden de volgende symptomen vaak gezien; pulmonale klachten zoals hoest en respiratoir falen (42%), lymfadenopathie (33%), neurologische symptomen zoals coma, delier en epilepsie mogelijk bij meningitis, bloeding, sinus cavernosus syndroom (25%), huidafwijkingen zoals oedeem, huiduitslag en panniculitis-like noduli (25%), gastrointestinale klachten zoals diarree, braken, buikpijn, bloedingen, ulceraties (18%), nierbetrokkenheid zoals insufficiëntie, nefrotisch syndroom (16%) en encephalopathie (9%).
| Diagnostiek |
|
Laboratorium onderzoek: Bellen met immulogielab (Esther van Leeuwen/ Paul Baars) om analyse soluble CD25 en NK cel activiteit in te zetten. |
|
Onderzoek naar een infectieus focus: Bellen met dienstdoende viroloog om spoed virale diagnostiek in te zetten (sein 58871), wanneer het sample voor 9 uur op het virologie lab is (zelf brengen) volgt er dezelfde dag (werkdagen) een uitslag |
| Onderzoek naar een maligniteit: CT hals, thorax, abdomen (zo nodig PET-CT) ter analyse van solide maligniteiten/ lymfomen + eventueel biopsie/punctie afwijkende massa Beenmerg aspiraat en biopt |
| Onderzoek naar betrokkenheid centraal zenuwstelsel: Liquor onderzoek (cel aantal, eiwit, glucose, lactaat, LDH) (cave stollingsstoornissen) MRI hersenen |
| Onderzoek naar een erfelijke vorm van HLH*: Genetische analyse bij alle HLH patiënten Risicogroepen primaire HLH: familie anamnese met consanguiniteit, partieel albinisme, jonge mannen met EBV/lymfoproliferatie. Functionele testen; NK-cel cytotoxiciteit en CD107 up-regulatie bij hoge verdenking erfelijke vorm |
Diagnose
De diagnose mag gesteld worden bij een moleculaire diagnose of als patiënten aan 5 van de onderstaande beschreven 8 criteria voldoen.4
| Diagnostische Criteria | Mechanisme |
| 1. koorts T> 38,5° | interleukines, TNF-α |
| 2. splenomegalie | infiltratie lymfocyten en histiocyten/macrofagen |
| 3. cytopenie (ten minste 2 cellijnen): Hb <5,6 mmol/l Neutrofielen <1x109/l, Trombocyten < 100x109/l |
suppressie door cytokines, hemofagocytose en downregulatie CD47 stamcellen |
| 4. hypertriglyceridemie (nuchter >3,0mmol/l) en/of hypofibrinogenemie (<1,5g/l) | plasminogeen-activator productie macrofagen lipoprotein lipase suppressie door cytokines |
| 5. hemofagocytose in beenmerg/milt/ lymfklier of lever | macrofaag activatie |
| 6. verlaagde of afwezige NK-cel activiteit | genetisch defect, tijdelijke dysfunctie door cytokine storm |
| 7. ferritine > 500ug/l | vrijkomen uit geactiveerde macrofagen en hepatocyten, |
| 8. soluble CD25 concentratie (> 2400 U/ml) | T-cel activatie |
Ook wordt vaak gezien: hyperbilirubinemie, hepatomegalie, transaminitis, verhoogd LDH, en een verhoogd d-dimeer (soms bij een normaal INR, APTT en fibrinogeen)2.
Behandeling:
Een uniform behandeladvies bij HLH is gezien de heterogeniteit van de onderliggende uitlokkende factoren niet te geven.1 Het doel is om de onderliggende trigger te behandelen en het overactieve immuunsysteem te remmen.
Stabiele patiënten zonder orgaanfalen
Bij een mild beloop/presentatie van HLH bij bv een infectie of een reumatologische aandoening kan behandeling van het onderliggende ziektebeeld voldoende zijn.
Bij HIV kan HAART worden gestart, bij EBV met een load van >10.000 copies wordt rituximab 375mg/m2 1 x per week gedurende 4 weken aangeraden. Bij een auto-immuun aandoening kan een behandeling met enkel corticosteroiden (methylprednisolon) voldoende zijn. Bij onvoldoende effect wordt Anakinra als tweedelijnsbehandeling gegeven, er is maar zelden etoposide nodig.1
Instabiele patiënten/orgaanfalen
HLH heeft echter frequent een zeer fulminant beloop en naast behandeling van de HLH-trigger is het tijdig starten van immunochemotherapie dan zeer belangrijk om de ongecontroleerde immuunactivatie te onderdrukken.
Patiënten worden behandeld volgens het HLH 1994-protocol, een pediatrische HLH-behandelrichtlijn. Deze bestaat uit een 8 weken durend schema met dexamethason en etoposide (150mg/m2) (2 weken inductie waarna 6 weken afbouwschema)4 Het HLH-2004 protocol, waarbij ciclosporine, in plaats van na 8 weken aan het begin van de behandeling werd gestart, bood geen significant overlevingsvoordeel. Etoposide wordt vanwege de pro-apoptotische werking op de geactiveerde cytotoxische T- en NK cellen gezien als een essentieel onderdeel van de HLH behandeling. Dosisreductie wordt niet geadviseerd bij cytopenieen. Wel dosis aanpassingen bij nier/lever dysfunctie volgens onderstaand advies bccancer.bc.ca.5
- dosisreductie van 25% geadviseerd bij een kreatinine klaring van 10-50ml/minuut, (onafhankelijk van billirubine)
- dosisreductie van 50% bij een klaring van <10ml/min (en een bilirubine < 50umol/L)
- dosisreductie van 75% bij een klaring van <10ml/min EN een verhoogd direct bilirubine van >50umol/L.
Bij een geïsoleerde bilirubinemie/verhoogde transaminasen is er in principe geen dosisreductie.1 Afwijkende leverenzymen zijn meestal in het kader van HLH activiteit in de lever. Bij oudere patiënten (>75 jaar) met een slechte performance status kan de etoposide dosering worden gereduceerd 25-50%.5
Inductietherapie voor HLH3
Dex = dexamethason, IT= intrathecaal, MTX= methotrexaat, HC= hydrocortisone.

Bij start HLH protocol:
- Inductie amenorrhoe bij menstruerende vrouwen (GnRH)
- Denk aan fertiliteitspreservatie
- Start schimmel, virale en PCP profylaxe
- HLA typering (voor evt stamceltransplantatie)
- ECG + echo cor (om uitgangswaarden te hebben)
- Overweeg IVIG bij infecties en hypogammaglobulinemie
In de follow-up adviseren we het ferritine als parameter van de ziekteactiviteit 3x per week te vervolgen. Het soluble CD25 1x per week (= de meest gevoelige parameter in de follow up)
Centraal zenuwstelsel betrokkenheid
Bij neurologische klachten, afwijkingen op MRI (niet aan een andere oorzaak toe te schrijven) of afwijkingen in de liquor die passen bij inflammatie is er mogelijk sprake van centraal zenuwstelstel (CZS) betrokkenheid (er is geen duidelijke definitie).6 Dit wordt met name bij kinderen beschreven.7 De behandeling bestaat uit intrathecaal methotrexaat (12mg) en hydrocortisone (15mg) 1 x per week. In het HLH94 schema wordt dit pas na 2 weken HLH behandeling gestart bij: toenemende neurologische klachten of niet verbeteren van het liquor onderzoek. Een occulte CZS lokalisatie (verhoogd eiwit of pleiocytose in de liquor zonder neurologische klachten) wordt ook behandeld. Omdat HLH bij volwassenen meestal secundair aan een onderliggende ziekte is, hoeft de onderliggende ziekte niet in het CZS te zitten. Het bewijs voor intrathecale behandeling is niet onomstotelijk sterk. In het HLH-94 protocol werd een deel van de patiënten behandeld met intrathecaal methotrexaat. Er werden geen verschillen gezien tussen de patiënten met CZS betrokkenheid die wel of niet intrathecaal behandeld werden.
Bij neurologische achteruitgang tijdens behandeling wordt geadviseerd een LP te verrichten en een MRI te maken. Denk aan bloeding, infectie, PRES of HLH in het CZS.
Patiënten met HLH hebben naast een verhoogd risico op bloedingen en infecties ook een verhoogd risico op PRES. Symptomen zijn hoofdpijn, verminderd bewustzijn, visus stoornissen en epilepsie. Er zijn geen specifieke testen maar het heeft karakteristieke afwijkingen bij MRI. Bij verdenking PRES: behandel hypertensie, streeft naar een normale vochtbalans/electrolieten en stop ciclosporine (als dit reeds gestart is). Eventuele intrathecale therapie wordt uitgesteld tot herstel PRES. Ook kan overwogen worden om bij ernstige PRES een dosis reductie te doen van het HLH behandelschema.
Recidief of refractaire ziekte
Bij een recidief na een eerdere goede respons of recidief tijdens de afbouwfase van het HLH94 protocol kan het HLH94 protocol worden herstart. Behandeling van refractaire ziekte is gebaseerd op case reports en kleine case series. ATG, Alemtuzumab, Anakinra, Ruxolitinib, emapalumab, DEP/CHOP-achtige schema’s, cytokine-adsorbtie kolommen, splenectomie zijn beschreven als opties.
Stamceltransplantatie
Bij refractaire ziekte, bewezen genetische oorzaak, centraal zenuwstelsel lokalisatie of een hematologische maligniteit (die niet kan worden genezen en dus een persisterende trigger kan zijn) bestaat er een indicatie voor een allogene stamceltransplantatie. Uitkomsten van stamceltransplantatie bij patiënten met nog actieve HLH zijn slechter gezien de cytokine storm en het risico op graft versus host. Bij maligniteit- geassocieerde HLH worden therapie schema’s voor de betreffende maligniteit gevolgd als pretransplantatie conditionering. Bij patiënten met een onderliggende genetische mutatie wordt in de aanloop naar een allogene stamceltransplantatie de HLH-94 therapie na de initiële 8 weken gecontinueerd met het onderhoudsschema (etoposide 150mg/m2 oraal wekelijks afgewisseld met dexamethason 10mg/m2 3 dagen).4 Belangrijk is om bij familiedonor eerst genetische diagnostiek in te zetten! Retrospectief onderzoek in kinderen toont aan dat een reduced-intensity conditioneringsschema de voorkeur heeft vanwege een lagere transplantatie gerelateerde mortaliteit. Een donorchimerisme van 20-30% wordt als voldoende beschouwd als bescherming tegen recidief HLH. Een flare van de HLH 2-3 weken post allogene stamceltransplantatie komt frequent voor en wordt behandeld met lage dosis etoposide/dexamethason.
Uit artikel La Rosee, blood 2019

Referenties
1. La Rosee P, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood 2019;133:2465-77.
2. Ramos-Casals M. et al. Adult haemophagocytic syndrome Lancet 2014; 383:1503-16.
3. Rosado F. et al. Hemophagocytic lymphohistiocytosis, an update on diagnosis and pathogenesis. Am J Clin Pathol 2013;139:713-27.
4. Bergsten E. et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood 2017; 130(25):2728-38.
5. http://www.bccancer.bc.ca/chemotherapy-protocols-site/Documents/Lymphoma...
6. Horne A, et al. How to Treat Involvement of the Central Nervous System in Hemophagocytic Lymphohistiocytosis? Curr Treat Options Neurol. 2017;19(1):3.
7. Schram A. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood 2015;125:2908-14.
Geldig 03-03-2020 versie 1
JHM-HFS-137